NGS One
새로운 미래를 열어가는 원오믹스
WGS
WGS는 유전체 전체를 광범위하게 읽어내어 분석하는 시퀀싱 방법으로 유전체 모든 영역에서 발생하는 유전적 변이를 찾아낼 수 있는 방법으로서, 타겟을 선별하지 않고 인트론(intron)과 비암호화 영역의 유전체(Noncoding region)를 함께 분석이 가능하여 구조적(structural) 변이나 유전자 발현 조절과 관련된 변이를 검출할 수 있는 장점이 있습니다.
인간이 갖고 있는 30억 개(genome) 뿐만 아니라 동식물과 미생물의 총 염기서열을 알아보기 위한 시퀀싱 방법으로서 1953년 왓슨, 크릭, 프랭클린이 DNA 구조를 발견한 후, 1965년 로버트 할리가 최초의 tRNA를 시퀀싱하여 1986년 노벨상을 수상하였고, 1977년에는 "chain termination method"이라고 하는 방사성 표지된 부분 소화 단편을 활용한 최초의 DNA 시퀀싱 방법을 개발하여, 이후 30년 동안 시퀀싱 세계를 지배하였고, 1998년 Solexa를 설립한 Shankar Balasubramanian과 David Klenerman은 형광 염료를 활용하는 새로운 “sequencing-by-synthesis method”을 개발하였고, 2005년은 Jonathan Rothberg와 동료들이 자동화 시스템에 pyrosequencing technology 기술을 구현한 454 시스템은 시장에 출시된 최초의 차세대 시퀀싱 플랫폼이었습니다.
Illumina가 2007년에 Solexa라는 회사를 인수하면서 세계에서 가장 널리 사용되는 NGS 기술을 제공하기 시작했으며 현재까지 NGS 플랫폼 시장의 선두 주자입니다.
◆ Data 분석
Mutation
Copy Number Alterations(CNA)
Structural Variants(SV)
◆ NGS Platform
NovaSeq 6000
NextSeq 500, MiSeq
PacBio RS II, PacBio Sequel
◆ Sample Requirements
Genomic DNA
Amount ≥200 𝑛𝑔
Volume ≥ 20 𝜇𝑙
Concentration : ≥ 10 𝑛𝑔/𝜇𝑙
WES
전장엑솜분석(whole-exome sequencing, WES)은 실제 단백질을 합성하는 부분인 엑손 부분만을 선별하여 염기서열을 분석하는 것으로 분석 부위는 총 30Mb 정도입니다.
전체 엑손들의 집합을 뜻하는 엑솜(exon + ome = exome)은 전체 유전체 중 약 1% 정도를 차지하는 약 3천만 개의 염기쌍으로 구성되어 있으나, 실제로 병을 일으키는 유전 변이들의 약 80% 정도가 이 엑솜에서 발견되며 엑솜 영역만 선택적으로 분석할 수 있기 때문에 효율적이고 비용 측면에서 경제적입니다.
또 특정 유전자의 변화가 질병과 직접적으로 연관되는 경우가 많기 때문에 엑솜 영역에서의 염기서열의 변화를 분석하는 것이 원인 유전자를 탐색하는 데 효과적일 수 있습니다.
또한 약 3천만 개의 염기서열 중에서 특정 질병과 관계가 있는 유전자만 선별하여 타겟 시퀀싱을 수행함으로써 보다 정확한 염기서열 정보 뿐만 아니라 저렴한 비용으로 정보를 확보 할 수 있습니다
◆ Data 분석
CNV (Copy Number Variation)
Various Variant Calling Pipeling
Cancer Analysis/ Family Analysis / Population Analysis
◆ NGS Platform
HiSeq 2500 / HiSeq 4000 / HiSeq X Ten / NovaSeq 6000
NextSeq 500
◆ Sample Requirements
Genomic DNA
Amount ≥200 𝑛𝑔
Volume ≥ 20 𝜇𝑙
Concentration : ≥ 10 𝑛𝑔/𝜇𝑙
RNA
DNA에 적혀 있는 유전정보를 mRNA로 옮기는 과정을 전사라 하며, 인간은 약 2만개 유전자가 확인되고 있으며 다양한 타입의 전사체가 발현되고 있다. 따라서 건강 및 질병 상태에서 보여지는 전사체 발현값을 분석함으로써 특정 유전자의 상태를 파악하는데 사용합니다.
RNA-sequencing은 transcriptome을 분석하여 RNA 발현의 차이와 돌연변이를 확인하는 분석방법입니다. Transcriptome은 전사체를 의미하는 transcript(ion)에 -ome이 붙은 말로 전사로 인해 만들어진 물질의 집합을 의미합니다. RNA read 정보를 분석하여 샘플에 존재하는 RNA의 종류 및 양을 확인하는 방법으로 발현 중인 유전자를 광범위하게 파악할 수 있습니다.
NGS를 이용하여 mRNA, Alternative splicing을 통한 다양한 전사체 분석, ncRNA(non-coding RNA)의 검사, fusion gene(융합유전자)를 검출할 수 있습니다.
RNA는 변성되기 쉽기 때문에 library 제작할 때에는 cDNA(complementary DNA)로 합성하는 과정이 필요하며, 이후 단계는 DNA 시퀀싱과 동일합니다.
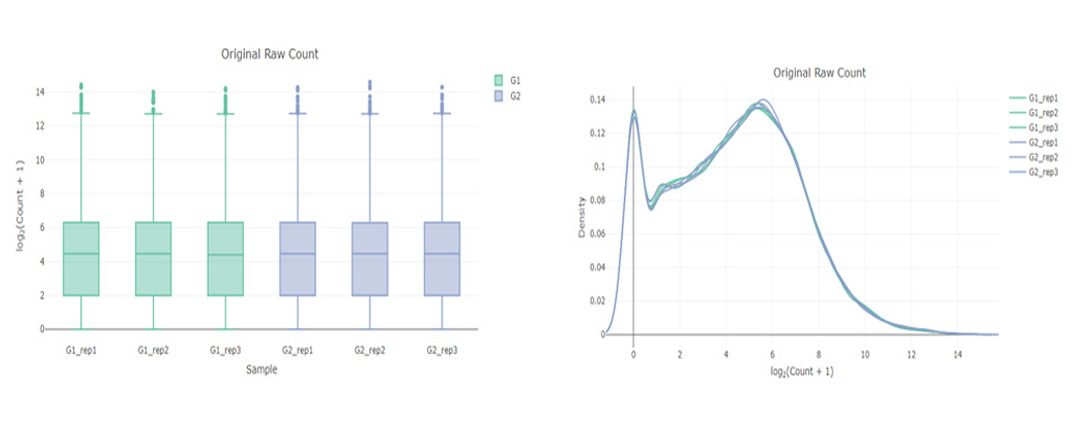
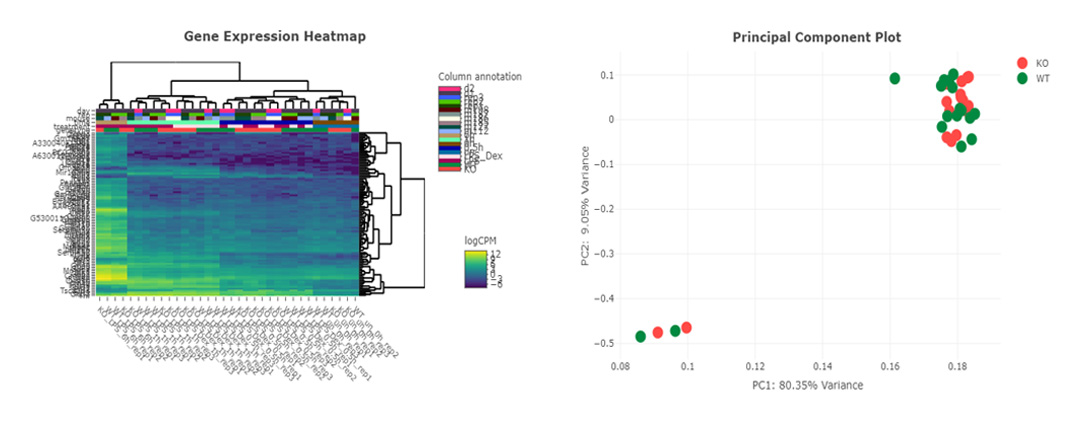
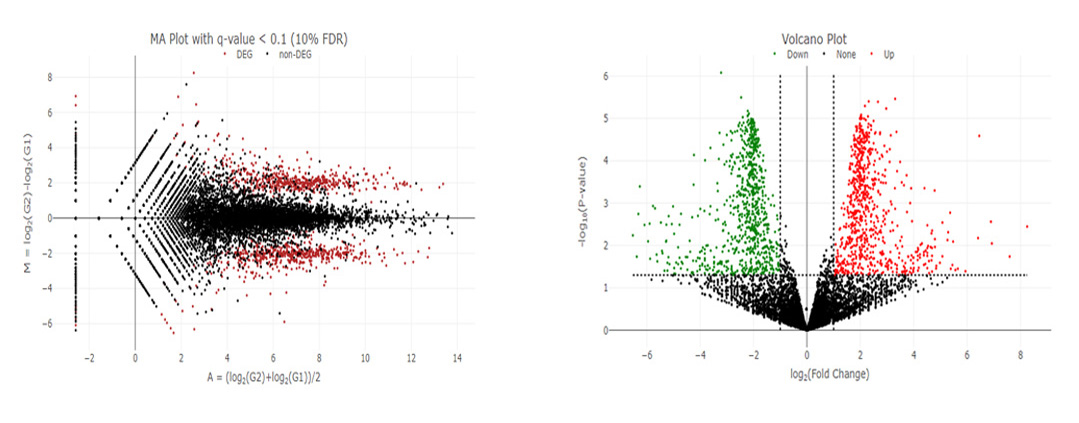
◆ Data 분석
mRNA Expression
Identification of miRNA
Target Prediction
multidimensional Scaling
Heatmap
Gene Set Enrichment Analysis
◆ NGS Platform
NovaSeq 6000
◆ Sample Requirements
Total RNA(Blood etc)
Amount ≥800 𝑛𝑔
Volume ≥ 20 𝜇𝑙
Concentration : ≥ 10 𝑛𝑔/𝜇𝑙
small RNA
Small RNA(siRNA)는 non-coding RNA 중 200bp 미만의 작은 크기의 RNA를 가리키며 유전자 발현에 중요한 역할을 하고 있는 것으로 알려져 있습니다.
그중 miRNA는 siRNA 중 하나로 약 21~24 bp의 크기로 유전자의 발현 조절에 있어서 중요한 역할을 하며 질병 유발 여부를 조정하는 상위 조절인자로서 중요한 연구대상이 됩니다.
micorRNA(miRNA)는 non-coding RNA 중 20bp 내외의 작은 크기의 RNA를 가리키며 유전자의 발현 조절에 있어서 중요한 역할을 하며 질병 유발 여부를 조정하는 상위 조절인자로서 중요한 연구대상이 됩니다.
◆ Data 분석
miRNA Expression & DEG
Identification of miRNA
Target Prediction
◆ NGS Platform
iSeq 100 / NextSeq 500 / NovaSeq 6000
◆ Sample Requirements
Total RNA (Blood etc)
Amount ≥2 𝜇𝑔
Volume ≥ 20 𝜇𝑙
Concentration : ≥ 50 𝑛𝑔/𝜇𝑙
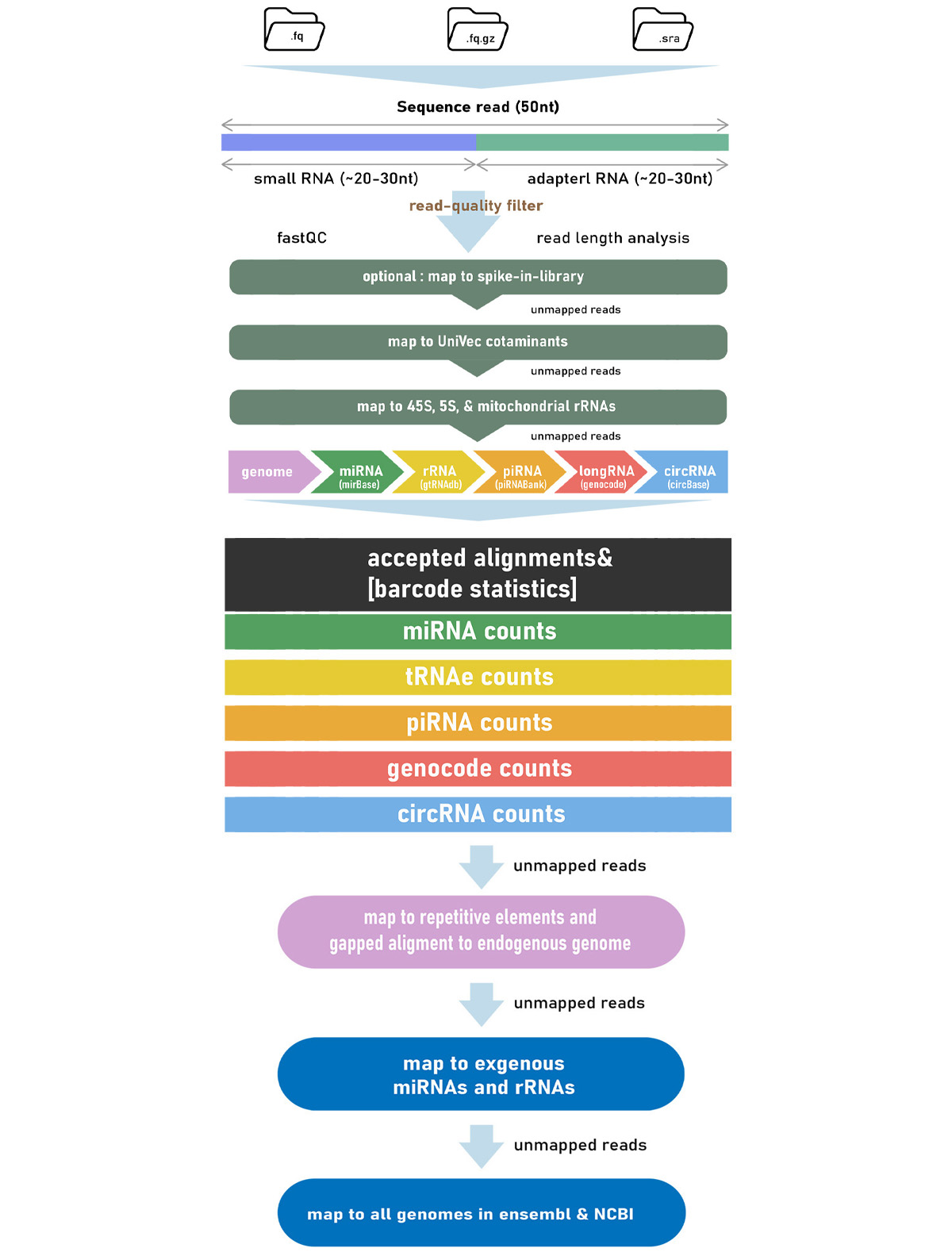
생물이 갖고 있는 고유의 DNA 정보가 바뀌지 않은 상태에서 유전자 작용을 조절하는것을 epigenetics(후성유전학)라고 하며,
DNA 메틸화, 히스톤 변형에 따른 RNA 조절을 통하여 유전자를 조절하게 됩니다.
DNA 메틸화는 주로 CpG 디뉴클레오티드 내에서 시토신 잔기의 5-탄소 위치에서 발생하여 5-메틸시토신(5-mC)을 형성하게 됩니다.
다시 말해서, DNA 메틸화는 DNA에 화학 그룹을 추가하고, 유전자를 "읽기" 위해 DNA에 부착되는 단백질을 차단하거나, 탈메틸화라는 과정을 통해 제거할 수 있습니다.
일반적으로 메틸화는 유전자를 “off" 탈메틸화는 유전자를 “on“ 줌으로써 유전자 발현을 조절하게 됩니다.
이러한 현상은 CpG 뉴클레오티드가 많이 모여있는 부분을 CpG island라고 이야기하는데 이 부분은 메틸화 현상이 없다가 암과 같은 질환이 발생할 때 급격하게 메틸화가
이루어져 많은 연구자들이 관심을 갖고 있는 부분입니다.
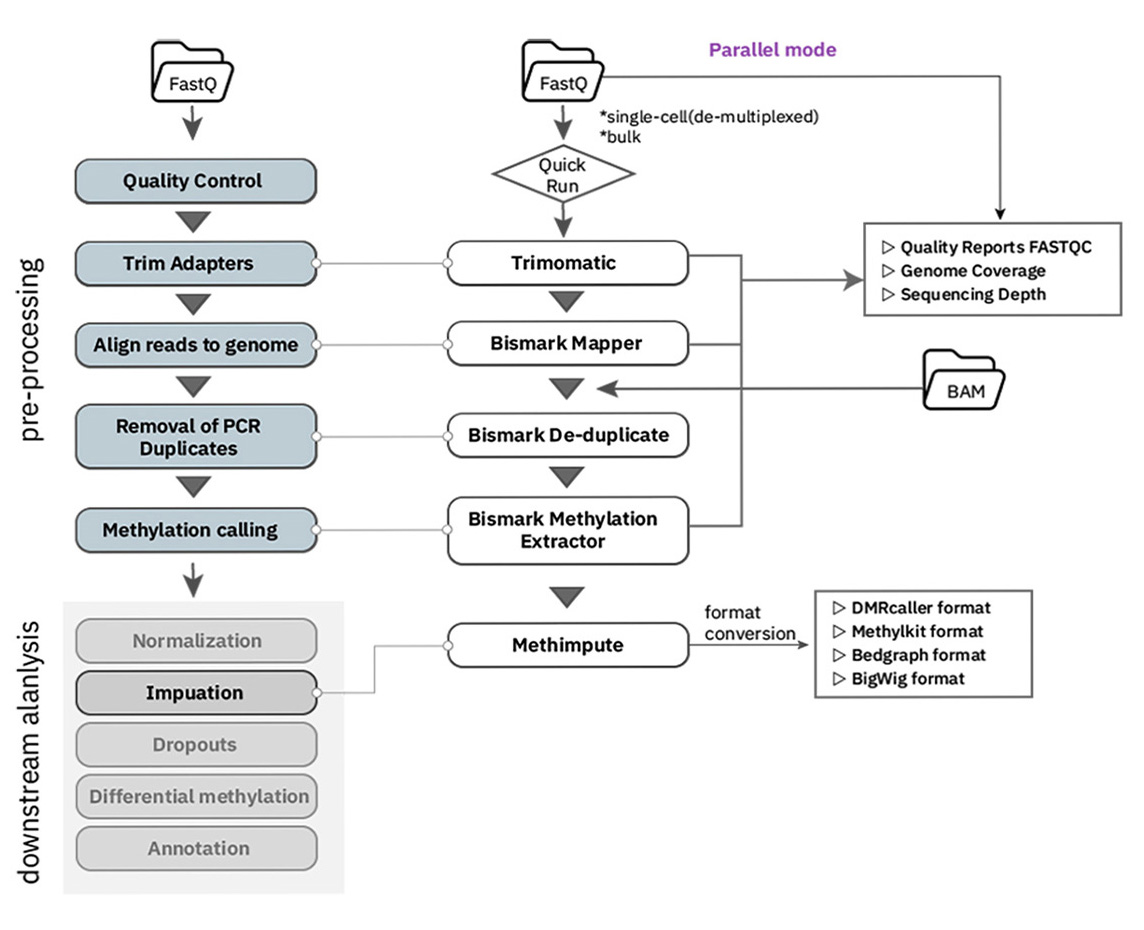
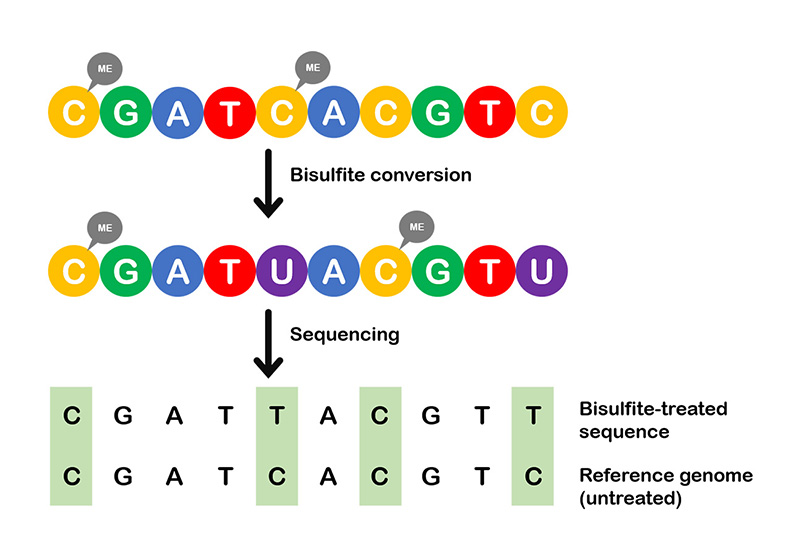
WGBS는 DNA에서 메틸화된 시토신을 검출하기 위한 방법으로 DNA에 bisulfite 처리를 하면, 메틸화되지 않은 시토신은 우라실로 변환한게 되며,
메틸화 시토신(5-mC)은 변환되지 않기 때문에 메틸화 시토신과 비 메틸화 시토신을 구별할 수 있는 원리를 이용하여 genomic DNA에 bisulfite 처리 후, 메틸화된 시토신의 위치를 NGS 시퀀싱을 통하여 구별할 수 있습니다.

Metagenome
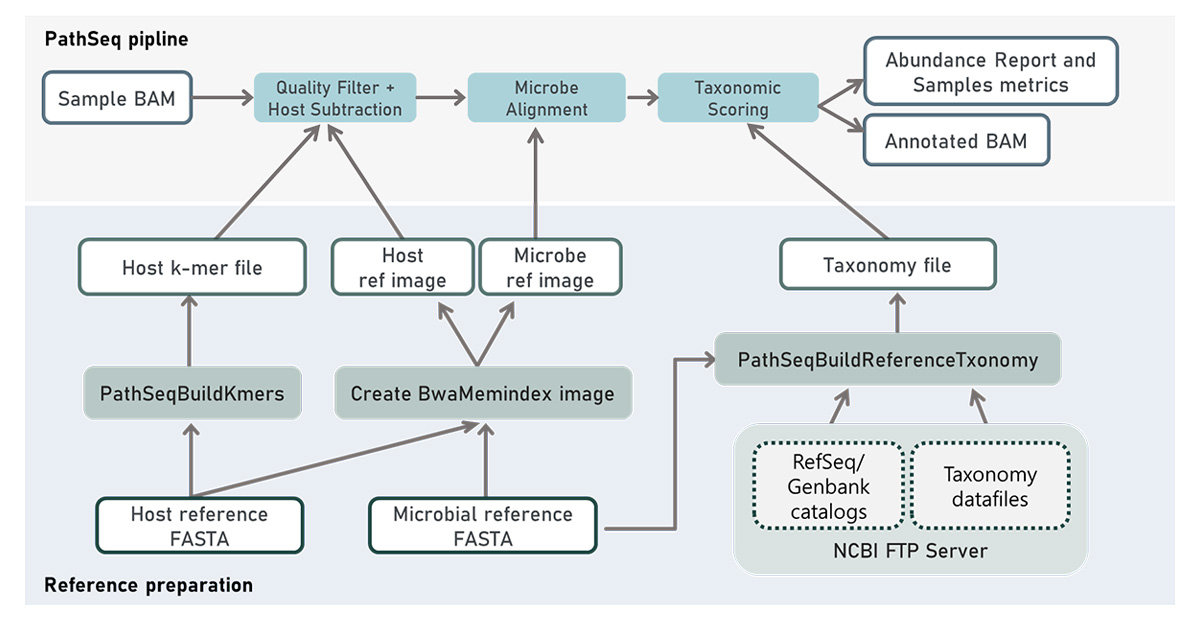
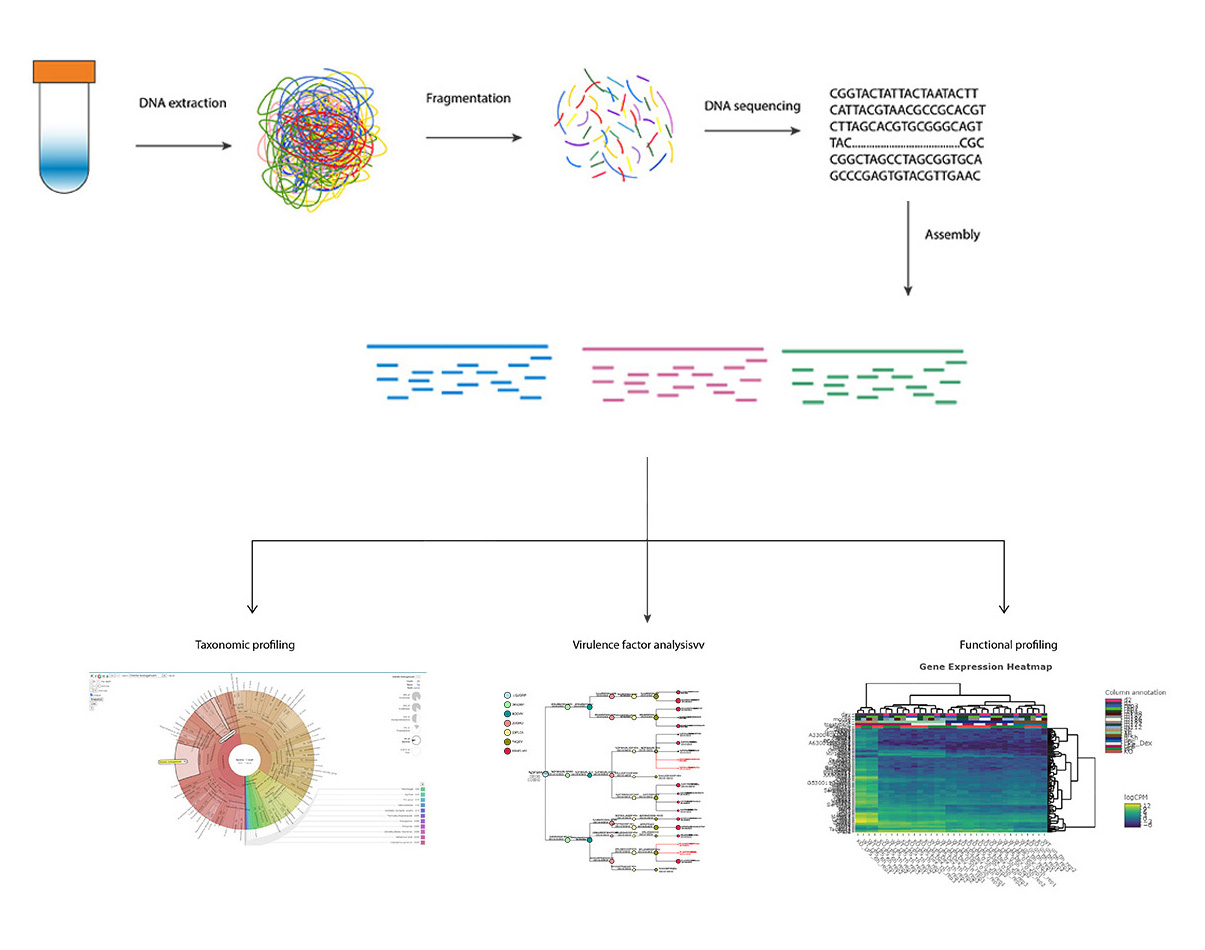
메타지놈(Metagenome)은 meta, gene, -ome이 합쳐진 말로(meta+gene+ome) 검체에 다양한 개체의 유전자가 많이 모여있는 것을 말합니다.
검체를 대상으로 NGS 플랫폼을 통하여 미생물(Microbial)의 염기서열을 읽어들여 미생물의 군집구조(microbial community) 및 메타유전체(metagenome)을 분석할 수 있습니다.
미생물을 직접 배양하지 않고 채취한 검체의 DNA를 통해 세균들의 유전정보를 획득하여 조성을 확인할 수 있으며, 이로부터 새로운 천연물, 항생제, 새로운 기능을 가진 생물활성 분자 등을 발견할 수 있습니다. 바이러스와 세균 군집을 분석하는 주요 방법으로 메타지놈 전체 유전자의 염기서열을 분석하는 샷건시퀀싱(shotgun sequencing), 특정 보존 영역(16s rRNA, 18s rRNA, ITS 영역)을 PCR 프라이머로 증폭시키고 NGS 장비를 이용한 amplicon sequencing이 있습니다.
◆ Data 분석
Phylogenetic Tree
Hierarchical Taxonomy Graph
Heatmap
PCA biplot
◆ NGS Platform
16S rDNA sequencing MiSeq, iSEQ system
- V3 to V4 Regions
- Cutomized Regions
Full-length 16s rRNA sequencing on PacBio RS II or Sequel
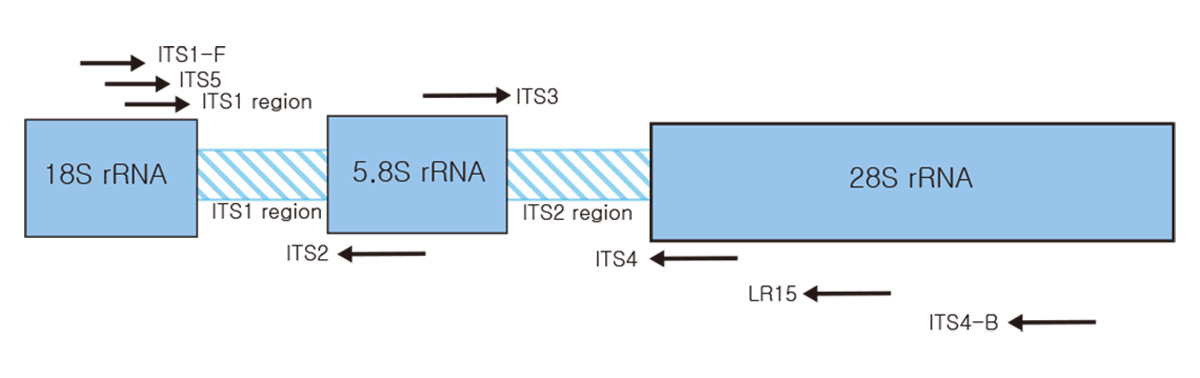
18S rDNA/ITS sequencing
◆ Sample Requirements
Genomic DNA
Amount ≥200 𝑛𝑔
Volume ≥ 20 𝜇𝑙
Concentration : ≥ 10 𝑛𝑔/𝜇𝑙
16S*18S*1TS
16S rRNA (10~18kb), 18S rRNA(총 길이 1,500-2,000 bp) 및 ITS(약 400-900 bp) microbiome 분석을 위한 amplicon full-length sequencing
OTU 클러스터링 및 필터링
OTU 분석 및 종 주석
PCA, 벤 다이어그램. OTU 풍부도를 기반으로 순위 곡선
알파 다양성, 베타 다양성, 메타 분석
다변수 통계 분석